
More Information
Submitted: July 06, 2023 | Approved: July 11, 2023 | Published: July 12, 2023
How to cite this article: Yaman M, Mustika S. Lung Abnormalities in Liver Cirrhosis. J Pulmonol Respir Res. 2023; 7: 015-020.
DOI: 10.29328/journal.jprr.1001045
Copyright License: © 2023 Yaman M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Cirrhosis hepatitis; Hepatopulmonary syndrome; Portopulmonary hypertension; Hepatic hydrothorax; Spontaneous bacterial empyema

Lung Abnormalities in Liver Cirrhosis
Muli Yaman1* and Syifa Mustika2
1Department of Pulmonology and Respiratory Medicine, Faculty of Medicine, Universitas Brawijaya, Malang, Indonesia
2Gastroentero-Hepatology Division, Department of Internal Medicine, Faculty of Medicine, Universitas Brawijaya, Malang, Indonesia
*Address for Correspondence: Muli Yaman, Department of Pulmonology and Respiratory Medicine, Faculty of Medicine, Universitas Brawijaya, Malang, Indonesia, Email: muliyaman@gmail.com
This article aims to provide what lung disorders can be caused by liver cirrhosis and also explain the pathophysiology of each etiologies. Regardless of preexisting lung illness, patients suffering from liver cirrhosis, especially decompensated liver cirrhosis can develop distinct pulmonary complications. Liver cirrhosis patients should be assessed for hepatopulmonary syndrome (HPS), portopulmonary hypertension (PoPH), hepatic hydrothorax (HH), and spontaneous bacterial empyema (SBEM) which are the most clinically significant pulmonary consequences, in particular when dyspnea develops in conjunction with hepatic cirrhosis. These entities differ in terms of pathophysiology, clinical characteristics, diagnosis, and suitable treatment options. This emphasizes the need for a specific diagnostic algorithm in liver cirrhosis patients presenting with dyspnea or other pulmonary symptoms. These pulmonary complications might be rare in patients with liver cirrhosis and portal hypertension but these complications might carry significant morbidity and mortality risks and, therefore, strong clinical suspicion is required to make an early accurate diagnosis. There are several medical therapies available for each condition in multiple studies but most of the treatments and procedures don’t have a significant benefit or have short-lived benefits. The only treatment that changes the clinical prognosis of decompensated cirrhosis effectively in the long term is liver transplantation. However, liver transplantation also needs careful consideration as in some cases it might increase the risk of morbidity and mortality.
Chronic liver diseases might further develop into cirrhosis. According to studies, within developed countries, Hepatitis C Virus (HCV), Hepatitis B Virus (HBV), alcoholic liver disease, and Non-alcoholic Steatohepatitis (NASH) are the most prevalent etiologies. Other plausible etiologies include alpha-1 antitrypsin deficiency, Budd-Chiari syndrome, Wilson disease, cirrhosis included by autoimmune hepatitis, primary biliary and primary sclerosing cholangitis, hemochromatosis, drug-induced cirrhosis of the liver, and chronic right heart failure [1].
It has been known for a while that chronic liver disease does coexist with changes in pulmonary function. It was already reported in 1977 on a postmortem examination of the lungs showing extensive vasodilatation of the pulmonary vasculature in patients suffering from liver cirrhosis. It was then suspected that these changes are related to the pulmonary clinical changes in patients suffering from chronic liver disease [2].
Patients with hepatic cirrhosis are at risk for developing respiratory problems. It is important to distinguish between these specific problems and primary lung conditions such as Chronic Obstructive Pulmonary Disease (COPD), which might also affect liver patients but isn’t linked to hepatic cirrhosis. Hepatopulmonary syndrome, portopulmonary hypertension, hepatic hydrothorax, and pulmonary empyema, are some of the most prevalent and clinically distinct pulmonary consequences [3]. Pulmonary complications increase the risk of further mortality and morbidity [4,5].
Liver cirrhosis: causes and pathophysiology
Cirrhosis can occur due to intoxication (alcoholism), infection (Hepatitis B, Hepatitis C), allergic reaction, immunopathological/ autoimmune disorder (autoimmune hepatitis, autoimmune cholangiopathy), or congenital metabolism disorders (inherited metabolic liver disease such as hemochromatosis, Wilson’s disease, cystic fibrosis, or a1 Antitrypsin deficiency). Worldwide, around 2 million deaths are contributed to liver disease, where 1 million of them are due to cirrhosis. In Indonesia, according to a Riskesdas survey done in 2013, Hepatitis B prevalence reaches 7.1% of the population. In Indonesia, the proportion of pregnant women with reactive HBsAg is 1.61% in 2021. In addition, there were around 820,000 deaths in 2019 due to Hepatitis B virus infection, mainly occurring through the development of cirrhosis and hepatocellular carcinoma [6-8].
Regardless of the possible etiologies, pathological characteristics consist of fibrosis development resulting in architectural distortion with formations of regenerative nodules. This fibrosis process will then gradually decrease hepatocellular mass, and function, and alter liver vasculari-zation. Fibrosis induction started with hepatic stellate cells activations, increasing collagen and other matrix extra-cellular [7].
There are several cells contributing to the progression of liver fibrosis. The primary cell type implicated in this process is hepatic stellate cells (HSCs). Due to the response of constant liver injury, HSC decreases the expression of genes including glial fibrillar acidic protein, PPARγ (peroxisome proliferator-activated receptor gamma), loses lipid droplets, and activates into myofibroblasts. The expression of fibrogenic genes such as collagen Type I and alpha-smooth muscle actin (-SMA) begins in myofibroblast. They multiply and go to the liver injury site, secreting ECM. Vascular endothelial growth factor (VEGF) which is directly associated with HSC proliferation is also released by myofibroblasts. Myofibroblasts and fibrogenic genes would then alter the contractile tone of smooth muscle cells, thus increasing the sinusoidal blood flow (around sinusoids and hepatic venules), which will lead to further vascular syndromes in liver cirrhosis [5].
Another important component is hepatocytes. Osteopontin, Transcriptional coactivator with PDZ-binding motif (TAZ), NADPH oxidase 4 (NOX4), Notch, and Indian Hedgehog are just a few of the fibrogenic factors that hepatocytes begin to produce after liver injury. Furthermore, damaged hepatocytes may discharge exosomes containing micro RNAs (miRNAs) that contribute to hepatic stellate cell (HSC) activation. However, in the absence of persistent inflammation, hepatocyte-derived fibrogenic factors would not cause liver fibrosis [9].
The next components are inflammatory cells and cytokines induced by chronic inflammation. Neutrophils, Kupffer cells (hepatic macrophages), Th17, and bone marrow-derived monocytes are promoting HSC initiation by inducing cytokines and growth factor productions. Liver macrophage, specifically Kupffer cells (KC) is a primary source of Transforming Growth Factor-β (TGF-β). TGF-β binds to its receptor in HSCs, activating myofibroblast and collagen Type I and III synthesis inducing fibrogenesis. KC also mediates liver inflammation and is thought to exacerbate liver injury and fibrosis, particularly because KC is continuously activated by DAMP (Damage Associated Molecular Pattern) released by dead hepatocytes in the late stages of the disease. Other fibrogenic cytokine secreted during liver injury includes CCL2 promoting HSCs initiation by recruiting monocyte-derived macrophage. There are also PDGF (Platelet-derived growth factor) signaling pathways that are important to HSC initiation [9,10].
Aside from that, Reactive Oxygen Species (ROS) also promote HSC activation. Kupffer cells, not only contribute to cytokines and chemokines production but also further contribute to the production of ROS. NADPH oxidase (NOX) promotes ROS production. ROS would then also contributes to the activation of HSCs and further contributes to the progression of fibrosis [9,11].
Liver cirrhosis and lung complication
Cirrhosis might cause portal hypertension which further might cause esophageal and gastric varices. Furthermore, decompensated cirrhosis might develop into several complications such as ascites, hepatic encephalopathy, and variceal bleeding [10,12].
Pulmonary complications can occur irrespective of the severity of the cirrhosis. There are several specific lung complications such as hepato-pulmonary syndrome (HPS), porto-pulmonary syndrome (PoPH), hepatic hydrothorax and spontaneous bacterial empyema [3].
Hepatopulmonary Syndrome (HPS): The definition of hepatopulmonary syndrome (HPS) is a decrease in arterial oxygen saturation due to dilated pulmonary vessels in portosystemic shunting or advanced to decompensated liver disease. HPS tends to develop into more severe liver disease. HPS in cirrhosis patients is also reported to double the mortality rates [2,3].
In HPS there are microvascular changes in the pulmonary arterial circulation, namely vasodilation and neoangiogenesis. Studies have shown that excessive production of pulmonary vasodilatory factors (nitrogen (NO), carbon monoxide (CO), and endothelin-1 (ET-1) contributes to pulmonary vasodilation Liver cirrhosis and portal hypertension increases ET-1 production by hepatic cells, inducing more eNOS activation and accumulation of monocytes. Activation of eNOS and iNOS elevates NO production inside pulmonary vasculature. Accumulating monocytes and monocyte-derived macrophages express iNOS and produce heme oxygenase-1, induce the production of CO, and contribute to vasodilatation. The macrophages and monocytes may accumulate in the lungs due to the translocation of intestinal bacteria and endotoxemia due to liver disease in the patient. These cells produce tumor necrosis factor-alpha (TNF-α) which will induce iNOS activation and progressively produce heme oxygenase which causes heme degeneration and CO release. Angiogenesis also plays an important role in the development of HPS and has been confirmed by studies showing that inhibition of angiogenesis enhances gas exchange abnormalities. Angiogenesis is initiated by circulating monocytes that produced and upregulates the Correlation of fractalkine (CX3CL1) and Vascular Endothelial Growth Factor (VEGF). Both vasodilatation and neoangiogenesis lead to perfusion and unaltered alveolar ventilation mismatch. Thus limiting right-left shunt, and alveolar-capillary diffusion, and resulting in hypoxia.
There are two types of HPS, defined by the location of the vasodilatation. Type 1st type of HPS has vasodilated vessels on precapillary levels, near the place where gas exchange is performed in the lungs. In this type of HPS, supplemental O2 can increase the Partial Pressure of Oxygen (PaO2). However, in the 2nd type of HPS, where larger vasodilatation caused arteriovenous to shunt away from gas exchange units, supplemental O2 is not beneficial [2,3].
In the early phase of HPS, patients are usually asymptomatic. Cirrhotic patients with new HPS may experience unspecified dyspnea that worsens with exertion, tachypnea, orthopnea, platypnea, cyanosis, diffuse telangiectasis (spider naevi) and clubbed fingers. Platypnoea is a form of dyspnoea that gets worsen when sitting or standing and is relieved by lying down. Meanwhile, orthodeoxia is a decrease in PaO2 of more than (upright) 5% or more than 4 mmHg when moving from supine to standing or sitting. Orthodeoxia is a result of increasing Ventilatory/Perfussion (V/Q) mismatch and decreasing cardiac output due to a shift from a supine to an upright position. Platypnoea and orthodeoxia are common features associated with HPS patients, although their sensitivity is low, they increase with the severity of HPS [2,3].
Pulse oximetry should be used as a first-line screening test. Mild hypoxemia have PaO2 of > 80 mmHg, moderate hypoxaemia have > 60 - < 80 mmHg PaO2, severe hypoxaemia have > 50 mmHg - < 60 mmHg PaO2, while very severe hypoxemia have < 50 mmHg PaO2. Pulse oximetry results below 96% for detecting HPS in patients with PaO2 below 70 mmHg are highly sensitive (100%) and specific (88%). The next testing in patients with suspected HPS is BGA. It is carried out within room air, with the patient seated first. This procedure is repeated in a standing position for around 15 to 20 minutes. Increase in AaDO2 (alveolar-arterial oxygen partial pressure difference) ≥ 15 mmHg (at the age younger than or equal to 64 years old) or ≥ 20 mmHg (at the age older than 64 years old). Orthodeoxia is characterized by an increase in PaO2 with 100% oxygen inhalation, which should be above 300 mmHg, and a reduction in PaO2 of 4 mmHg or 5% from supine to upright position [2,3].
Although a chest X-ray may show strong pulmonary vascular signs in the lower lobes, HPS is not always the cause of this finding. CT is also usually done only to exclude possible pulmonary pathologies. To rule out any other related intrinsic lung diseases, pulmonary function tests should be carried out. The test with the highest sensitivity for showing an intrapulmonary shunt is echocardiography with contrast. In order to create bubbles larger than 10-15 microns in diameter, agitated 0.9% saline or indocyanine green is intravenously injected during echocardiography. This is now established as the gold-standard method for evaluating intrapulmonary vasodilations. The test is positive when left atrial opacification is found with microbubbles between the 4th to 6th cardiac cycle. This can further be graded into stages 1 (less than 30 microbubbles), 2 (30 to 100 microbubbles), and 3 (more than 100 microbubbles). Other than the “positive” finding on contrast-enhanced trans-thoracic echocardiography, an abnormal brain uptake (larger than 6%) after 99mTcMAA (technetium-99m macro-aggregated albumin) 20-50 µm size lung perfusion scanning is also another reliable method to assess intrapulmonary vasodilation. Pulmonary angiography can also be used to differentiate type I and type II HPS. Type I HPS have a normal or “spongy” appearance, while type II has discrete arteriovenous communications [2-4].
Oxygen therapy is recommended for patients with severe hypoxemia (PaO2 ≤ 55 mmHg or SaO2 ≤ 88%), and given until liver transplantation can be performed. Reduction in hypoxemia might lead to better exercise tolerance and improved quality of life by decreasing symptoms of intrapulmonary vascular shunts. Many pharmacological therapies have not had significant outcomes according to several studies. Anti-angiogenic (somatostatin and sorafenib analogs), methylene blue, pentoxifylline, and anti-diabetes (metformin and pioglitazone) have not had a significant effect. Transjugular intrahepatic portosystemic shunt (TIPS) still can’t be recommended since there are still limited data with variation outcomes. It may exacerbate the hyperkinetic circulatory state, increase intrapulmonary vasodilatation, and shunting, exacerbate hypoxemia, risk of decompensation, and encephalopathy. The only recommended effective definitive treatment available is a liver transplant. This is the only method shown to give significant benefit to increase survival rate and increase quality of life. Coil embolization might improve HP’s persistent hypoxemia, both before and after liver transplant, but its use is limited in cases where there are large AV communications [2-4,8] Figure 1.
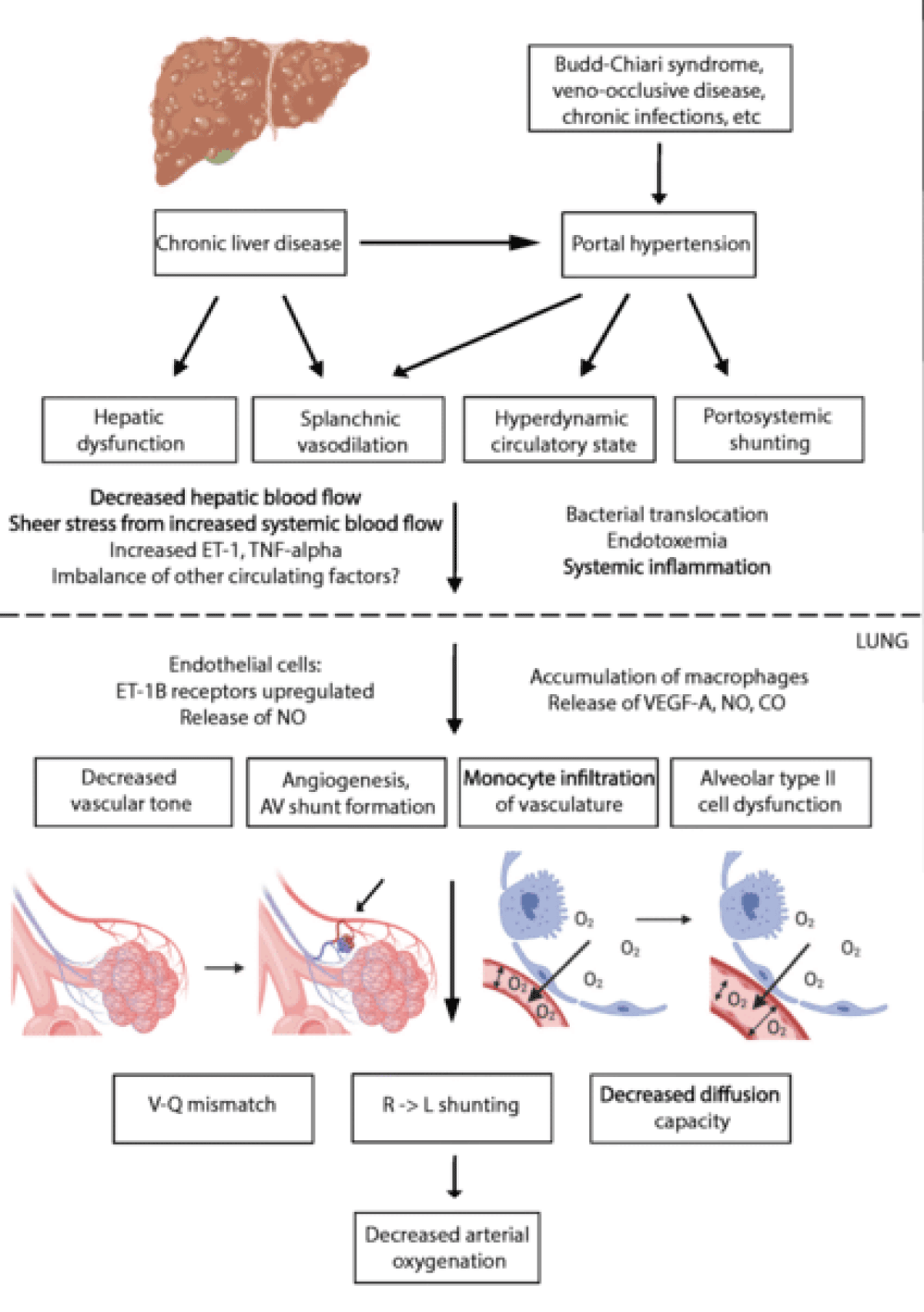
Figure 1: Pathogenesis of Hepatopulmonary Syndrome (HPS) [13]. A decrease in vascular tone, angiogenesis, and monocyte infiltration will contribute to V-Q mismatch, right-to-left shunting, and a decrease in diffusion capacity. These will further decrease arterial oxygenation.
Portopulmonal Hypertension (PoPH): Portopulmonary hypertension is defined as a disorder present with pulmonary artery hypertension (> 25 mmHg) during resting coupled with the presence of portal hypertension and pulmonary capillary wedge pressure ≤ 15 mmHg with or without significant liver disease. This condition has the same histological characteristic of plexogenic arteriopathy of idiopathic pulmonary hypertension. It also involves the proliferation of endothelial and smooth muscle [3,8].
Although its pathogenesis is still unknown due to a lack of animal models, some pathophysiological hypotheses were suggested. Firstly is the imbalance among vasoconstrictor and vasodilator mediators such as ET-1, thromboxane, IL-1, IL-6, and angiotensin. Hyperdinamic pulmonary circulation due to splanchnic vasodilation and increasing resistance to hepatic blood flow can also occur, resulting in portal venous hypertension. This will then increase sheer stress on the pulmonary vascular wall as a result of increasing turbulence, leading to vascular remodeling. Permanent vascular remodeling due to this damage to pulmonary endothelium and the underlying smooth muscle then occurs mediated by E2, bone morphogenetic protein 9 (BMP9), and Bone Morphogenetic Protein Receptor Type 2 (BMPR2). There’s also a hypothesis suggesting there might be elevated local inflammation and oxidative and nitrative stress as a result of increasing cytokine associated with liver cirrhosis. At the same time, portosystemic shunts and the inability of the liver to filter blood adequately from the digestive tract might result in the bypass of bacteria endotoxins and increasing vasoactive substance into the pulmonary circulation. Genetic polymorphism also has some role in disease progression. Although all the hypothesis above might seem similar to HPS, PoPH’s main pathophysiology is vasoconstriction, not vasodilatation [3,4,14,15].
PoPH is usually asymptomatic at first. History of risk factors must be assessed during history taking, including diseases associated with PH. Manifestations might occur from underlying liver disease or other complications and thus can be confused with PoPH manifestations itself, such as fatigue, weakness, orthopnea, or hemoptysis. History that must be assessed on patients suspected of PoPH is dyspnea both at rest or on exertion, weakness, fatigue, orthopnea, palpitations, pre-syncope, syncope, and chest pain. Cyanosis is rarely present. Physical examination might show protrusion of pulmonary component from P2 (second heart sound), right-sided S3, and a right-sided S4 on the right side. Tricuspid regurgitation murmur might also be present. Distended jugular venous, ascites, or edema on both lower extremities can also be found. Definitive diagnosis should be made by right heart catheterization and measuring Mean Pulmonary Arterial Pressure (MPAP), Pulmonary Artery Occluded Pressure (PAOP), Cardiac Output (CO), and Pulmonary vascular resistance (PVR) [3,4,14].
Portopulmonary hypertension staging is measured made based on mean pulmonary arterial pressure either by right heart catheterization (the gold standard method) or estimated cardio graphically (a front-line non-invasive alternative). MPAP of 25-35 mmHg is considered mild, 35 mmHg - 45 mmHg is moderate, and lastly > 45 mmHg is considered severe. Pulmonary arterial wedge pressure (PAWP) should be ≤ 15 mmHg, peripheral vascular resistance (PVR) should be > 240 dyn.sec.cm-5, and transpulmonary gradient (TPG) of > 12 mmHg coupled with PH clinical evidence. Meanwhile, an echocardiograph can predict RVSP by measuring peak TRV using the modified version of the Bernoulli equation. TRV > 3.4 m/sec or 2.9 to 3.4 m/sec along with echocardiographic findings of PoPH confirms a high possibility of PoPH. Using echocardiographic, estimated RVSP ≥ 35 mmHg usually implies Pulmonary arterial pressure (PAP) > 24 mmHg, while right ventricular systolic pressure (RSVP) < 30 mmHg can exclude PoPH. Electrocardiography might also show right atrial enlargement, right ventricular hypertrophy, Right Bundle Branch Block (RBBB), and deviated axis to the right [4,14].
Therapy for PoPH patients aims to reduce portal hypertension and prevent further complications (e.g., thrombo-embolism or right heart failure). Routine anti-coagulant administration is not recommended, as it increases the possibility of coagulopathy or esophageal varices, thrombo-cytopenia, and increased bleeding risk. Some of the available medications commonly used include endothelin receptor antagonists, prostacyclin pathway agonists, and Nitric Oxide (NO)-cyclic guanosine monophosphate enhancers (PDE5 inhibitors, such as sildenafil and tadalafil). CCB (calcium canal blockers) are not beneficial and BB (beta blocker) should also be avoided due to its side effect on increasing pulmonary resistance (PVR) and reducing right ventricle cardiac output [3].
Liver transplantation (LT) in PoPH patients are complex as not all patient would benefit from LT. Post-LT outcomes in PoPH patients can be unpredictable and worsening pulmonary hypertension might occur, increasing the mortality rate [3,16].
In a cohort done on 228 patients, higher PVR before LT is associated with worse outcomes. In this study, PoPH survival post-LT was modest despite effective PA-targeted therapy [17]. An article reviewing current PoPH therapy and LT in Japan uses the indication of MPAP < 35 mmHg and PAVS < 400 dyn/s/cm-5 as an indication for LT, regardless of therapeutic drug usage [18]. However one study suggests that MPAP > 35 mmHg and PVR < 250 dynes/s/cm-5 with preserved RV function might survive LT [19].
In conclusion, LT might improve PH and be effective to treat PoPH, but it still needs PAH-specific therapy, otherwise, poor prognosis post-LT might still be found [20] Figure 2.
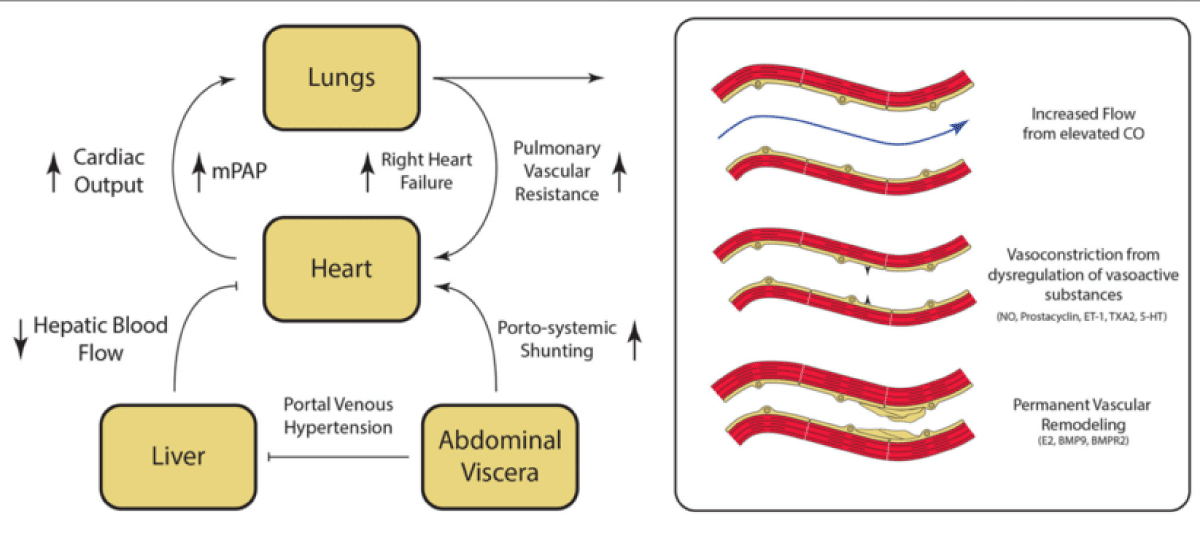
Figure 2: Pathogenesis of Portopulmonary Hypertension (PoPH) [14] Fibrosis on the liver induces PH, followed by splanchnic vasodilatation and haptic blood flow resistance to increase. The overall circulating volume will then increase and blood flow diversion from the liver to the heart through portosystemic shunting will lead to a hyperdynamic state. At the same time, remodeling of the vasculature mediated by inflammatory factors and cytokines will contribute to PH development.
Hepatic Hydrothorax (HH): Pleural effusion, typically > 500 mL in liver cirrhosis patients who has no coexisting cardiac or pulmonary disorders is referred to as hepatic hydrothorax (HH). This condition is thought to affect 5% - 10% of patients with cirrhosis. The exact pathophysiology is not completely understood yet, but there are some proposed hypotheses. Currently, trans diaphragmatic fluid shift to the pleural cavity through pleuroperitoneal communications from the peritoneal is the favored hypothesis. These might be observed in patients present with diaphragmatic lesions, frequently on the right hemidiaphragm. This is because, compared to the left side, the right side is less muscular and thinner [3,4,21].
Even with a tiny amount of pleural effusion, patients with restrictive patterns of pulmonary function can nevertheless experience severe clinical symptoms. Patients may experience dizziness, fatigue, dyspnea at rest, dyspnea with exertion, pleuritic chest pain, chest discomfort, or non-productive cough. However clinical manifestation won’t be specific, since usually HH coexists with ascites or other features of PH. Symptoms vary further according to effusion volume, rapidity of accumulation, and associated cardiopulmonary disease presence [3,21].
Diagnosis is performed based on thoracocentesis, distinguishing transudate and exudate. Pleural fluid analysis of HH will show the nature of transudative effusion with a similar feature to ascetic fluid. Total PMN (Polymorphonuclear) cell count should be < 250/µL, total protein concentration < 2.5 g/dL, a serum-to-pleural-albumin gradient > 1.1 mg/dL, or an albumin quotient (pleural fluid/serum) < 0.6. LDH gradient < 0.6 (serum-pleural fluid), protein quotient < 0.5 (pleural fluid/serum), pH value of 7.4 to 7.55, and pleural glucose level similar to serum level. Further imaging diagnoses such as Ultrasonography (USG) and chest X-ray are valid to rule out other pulmonary diseases and malignancies. In some cases, 99mTc-human serum albumin might also confirm HH when radioisotopes migrate into pleural space from the peritoneal cavity [3,4,21].
Liver transplantation remains the best choice for decompensated cirrhosis. It is also shown to provide the best long-term survival and should be considered in all patients. On patients not eligible to perform LT, other procedures can be considered. Thoracocentesis is effective in the relief of symptoms, although the benefits are short-lived thus procedure needs to be repeated. TIPS can also be performed especially on refractory HH. TIPS is also superior compared to other modalities on rebleeding from varices prevention, however, it doesn’t improve end-stage liver disease prognosis. Medical management involves eliminating and preventing ascites’ recurrence. This includes a sodium-restricted diet (70-90 mmol/day), weight loss of 0.5 kg/day in non-edematous patients and 1 kg/day in edematous patients. Spironolactone 100 mg/day and loop diuretics such as furosemide 40 mg/day are used as the initial regimen to excrete renal sodium > 120 mEq/day. Medication dose may be increased every 3-5 days up to 160 mg/day for Furosemide and 400 mg/day for Spironolactone [21].
Spontaneous bacterial empyema (SBEM): Spontaneous bacterial empyema is a spontaneous infection from a preexisting HH. This rarely occurs but needs to be considered. Diagnosis is based on total PMN (Polymorphonuclear) cell count < 250/mm3 with positive cultures or PMN > 500 cells/mm3 with negative cultures. Therapy consists of IV 3rd generation of Cephalosporins (2 g Ceftriaxone every 24 hours for 7-10 days). Piperacillin/Tazobactam or Carbapenem should be considered in countries with high antibiotic resistance [3].
Liver cirrhosis may rarely develop into several pulmonary complications. These complications may result in significant morbidity and mortality if not treated early on. Pulmonary complications might be suspected when dyspnea occurs in patients with cirrhosis. Several diseases that should be suspected include hepatopulmonary syndrome (HPS), porto-pulmonary hypertension (PoPH), hepatic hydrothorax, and spontaneous bacterial empyema which represent the most clinically relevant pulmonary complications of cirrhosis of the liver. Different diagnostic procedures should be performed personalized based on each manifestation. Patients with these illnesses should be examined and evaluated for liver transplantation eligibility since it is the only effective treatment that improves the clinical prognosis significantly.
- Sharma B, John S. Hepatic cirrhosis. 2018.
- Bansal K, Gore M, Mittal S. Hepatopulmonary Syndrome. 2020.
- Benz F, Mohr R, Tacke F, Roderburg C. Pulmonary Complications in Patients with Liver Cirrhosis. J Transl Int Med. 2020 Sep 25;8(3):150-158. doi: 10.2478/jtim-2020-0024. PMID: 33062591; PMCID: PMC7534492.
- Soulaidopoulos S, Goulis I, Cholongitas E. Pulmonary manifestations of chronic liver disease: a comprehensive review. Ann Gastroenterol. 2020 May-Jun;33(3):237-249. doi: 10.20524/aog.2020.0474. Epub 2020 Mar 27. PMID: 32382226; PMCID: PMC7196609.
- Shenoda B, Boselli J. Vascular syndromes in liver cirrhosis. Clin J Gastroenterol. 2019 Oct;12(5):387-397. doi: 10.1007/s12328-019-00956-0. Epub 2019 Apr 12. PMID: 30980261.
- Minister of Health of the Republic of Indonesia. (2023) Decree of the Minister of Health of the Republic of Indonesia Number HK.01.07/MENKES/15/2023. Decree of the Minister of Health of the Republic of Indonesia
- Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J. Medicina Interna de Harrison-2 Volumes-20. McGraw Hill Brasil. 2019.
- Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet. 2021 Oct 9;398(10308):1359-1376. doi: 10.1016/S0140-6736(21)01374-X. Epub 2021 Sep 17. PMID: 34543610..
- Gabbia D, Carpi S, Sarcognato S, Zanotto I, Sayaf K, Colognesi M, Polini B, Digiacomo M, Macchia M, Nieri P, Carrara M, Cazzagon N, Russo FP, Guido M, De Martin S. The phenolic compounds tyrosol and hydroxytyrosol counteract liver fibrogenesis via the transcriptional modulation of NADPH oxidases and oxidative stress-related miRNAs. Biomed Pharmacother. 2023 Jan;157:114014. doi: 10.1016/j.biopha.2022.114014. Epub 2022 Nov 12. PMID: 36379119.
- Engelmann C, Clària J, Szabo G, Bosch J, Bernardi M. Pathophysiology of decompensated cirrhosis: Portal hypertension, circulatory dysfunction, inflammation, metabolism and mitochondrial dysfunction. J Hepatol. 2021 Jul;75 Suppl 1(Suppl 1):S49-S66. doi: 10.1016/j.jhep.2021.01.002. PMID: 34039492; PMCID: PMC9272511.
- Slevin E, Baiocchi L, Wu N, Ekser B, Sato K, Lin E, Ceci L, Chen L, Lorenzo SR, Xu W, Kyritsi K, Meadows V, Zhou T, Kundu D, Han Y, Kennedy L, Glaser S, Francis H, Alpini G, Meng F. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am J Pathol. 2020 Nov;190(11):2185-2193. doi: 10.1016/j.ajpath.2020.08.014. Epub 2020 Sep 11. PMID: 32919978; PMCID: PMC7587925.
- Cates AL, Farmer B. Chronic Drug Use and Abdominal Pain. Emerg Med Clin North Am. 2021 Nov;39(4):821-837. doi: 10.1016/j.emc.2021.07.006. Epub 2021 Sep 9. PMID: 34600640.
- Iqbal S, Smith KA, Khungar V. Hepatopulmonary Syndrome and Portopulmonary Hypertension: Implications for Liver Transplantation. Clin Chest Med. 2017 Dec;38(4):785-795. doi: 10.1016/j.ccm.2017.08.002. Epub 2017 Sep 23. PMID: 29128026.
- Thomas C, Glinskii V, de Jesus Perez V, Sahay S. Portopulmonary Hypertension: From Bench to Bedside. Front Med (Lausanne). 2020 Nov 3;7:569413. doi: 10.3389/fmed.2020.569413. PMID: 33224960; PMCID: PMC7670077.
- Matyas C, Haskó G, Liaudet L, Trojnar E, Pacher P. Interplay of cardiovascular mediators, oxidative stress and inflammation in liver disease and its complications. Nat Rev Cardiol. 2021 Feb;18(2):117-135. doi: 10.1038/s41569-020-0433-5. Epub 2020 Sep 30. PMID: 32999450.
- Raevens S, Boret M, De Pauw M, Fallon MB, Van Vlierberghe H. Pulmonary Abnormalities in Liver Disease: Relevance to Transplantation and Outcome. Hepatology. 2021 Sep;74(3):1674-1686. doi: 10.1002/hep.31770. Epub 2021 May 24. PMID: 33636019.
- Cartin-Ceba R, Burger C, Swanson K, Vargas H, Aqel B, Keaveny AP, Heimbach J, Taner T, Nyberg S, Rosen C, Cajigas H, DuBrock H, Krowka MJ. Clinical Outcomes After Liver Transplantation in Patients With Portopulmonary Hypertension. Transplantation. 2021 Oct 1;105(10):2283-2290. doi: 10.1097/TP.0000000000003490. PMID: 33065725.
- Tokushige K, Kogiso T, Egawa H. Current Therapy and Liver Transplantation for Portopulmonary Hypertension in Japan. J Clin Med. 2023 Jan 10;12(2):562. doi: 10.3390/jcm12020562. PMID: 36675490; PMCID: PMC9867251.
- DuBrock HM, Runo JR, Sadd CJ, Burger CD, Cartin-Ceba R, Rosen CB, Taner T, Nyberg SL, Heimbach JK, Findlay JY, Krowka MJ. Outcomes of Liver Transplantation in Treated Portopulmonary Hypertension Patients With a Mean Pulmonary Arterial Pressure ≥35 mm Hg. Transplant Direct. 2020 Nov 10;6(12):e630. doi: 10.1097/TXD.0000000000001085. PMID: 33204828; PMCID: PMC7665265.
- Li J, Zhuang Q, Zhang X, Zheng Y, Qiao Z, Zhang J, Shen X, Shen J. Prevalence and Prognosis of Portopulmonary Hypertension in 223 Liver Transplant Recipients. Can Respir J. 2018 Sep 18;2018:9629570. doi: 10.1155/2018/9629570. PMID: 30319722; PMCID: PMC6167565.
- Lv Y, Han G, Fan D. Hepatic Hydrothorax. Ann Hepatol. 2018 January-February;17(1):33-46. doi: 10.5604/01.3001.0010.7533. PMID: 29311408.