
More Information
Submitted: October 31, 2022 | Approved: November 24, 2022 | Published: November 25, 2022
How to cite this article: Chaudhry IA, Khan MN, Alqahtani YA, Alghamdi A, AlFraih OM, et al. Pulmonary congenital cystic adenomatoid malformation: a rare congenital abnormality in adults and review of literature. J Pulmonol Respir Res. 2022; 6: 016-019.
DOI: 10.29328/journal.jprr.1001038
Copyright License: © 2022 Chaudhry IA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Congenital cystic adenomatoid; Malformation; Malignancy; Respiratory tract infection; Surgery

Pulmonary congenital cystic adenomatoid malformation: a rare congenital abnormality in adults and review of literature
Iftikhar Ahmed Chaudhry1*, Mohammad Nasim Khan1, Yousif A Alqahtani2, Abdullah Alghamdi2, Othman M AlFraih2, Meenal A AlAbdulhai2 and Ikram Ul-Haq Chaudhry2
1Medical College, Umm Al-Qura University, Makkah, Saudi Arabia
2Dammam Medical Complex, Saudi Arabia
*Address for Correspondence: Iftikhar Ahmed Chaudhry, Medical College Umm Al-Qura University, Makkah, Saudi Arabia, Email: [email protected]
Congenital cystic adenomatoid malformation of the lung (CCAM) is characterized by an adenomatoid proliferation of bronchiole-like structures and cysts formation. The condition is most commonly found in newborns and children and may be associated with other malformations; rarely, the presentation is delayed until adulthood. We herein report two cases of CCAM in adult patients. 22 years old healthy female with pre-employment health screening chest X-ray showed a lesion in the upper lobe of the right lung. In another case, a computed tomographic scan of the thorax (CT) confirmed a mass in the upper right lung. A 28-year-old male presented with recurrent respiratory tract infection resistant to antimicrobial therapy. CT scan of the thorax showed a mass in the left lung upper zone. Surgical resection was performed in both cases, and histopathology of the resected specimen showed both cases were consistent with the CCAM.
Congenital cystic adenomatoid lung malformation (CCAM) is a rare developmental anomaly known as unilateral dysplasia of the lung. It was described in 1787 as the absence of the lung. Ch’in and Tang first reported it in 1949 [1,2]. The development of the respiratory system begins at three weeks of gestation. There is a hypothesis that CCAM occurs due to the arrest of the development of the bronchial tree or due to alteration in bronchial development. Stocker, in 1977 classified CCAM into three types, widely used until 2002, then new reclassification of CCAM was reported with five subtypes [3,4]. 85% of the cases are diagnosed within the first two years of life and are rarely encountered in adults. Primarily it is interpreted as an incidental finding on radiological examination of chest X-Ray and CT scan of the chest. In adults, this usually involves one lobe, and the patient presents with a persistent cough, hemoptysis, recurrent respiratory tract infection, and lung abscess. Up to 26% of the cases of CCAM were associated with anomalies [5]. This case series is reported in line with PROCESS criteria [6].
Case 1
Twenty-four years old female was referred to us from the employee health clinic with opacity in the upper zone right lung. She had no medical illness in the past. Clinical examination of the neck, chest and abdomen was normal. There was no palpable lymphadenopathy. Basic blood investigations CBC, Liver and renal panels were normal. CT scan of the chest showed a mass in the right upper lobe. Mass was approached during the Right posterolateral thoracotomy and we found a hard mass in the apical segment of the upper lobe of the right lung. A segmentectomy was performed, and the patient was extubated on the table and had an uneventful recovery. The histopathology report showed a typical alveolar structure with multiple cysts and fibrovascular connective tissues. A Follow-up CT scan after four years was normal.
Case 2
A 26-year-old male presented to us with a history of cough and recurrent chest infection for the last six months, which did not resolve with antibiotics. He had no medical illness in the past, and basic blood investigations were normal. A chest X-Ray and CT scan of the chest showed a 5 cm × 3.5 cm mass in the upper lobe of the left lung. This was approached through a left posterolateral thoracotomy, and we found a large cystic mass in the left upper lobe with dense adhesions to the chest wall and pericardium. A left upper lobectomy was performed. The patient was extubated on the table and transferred to the ward. His post-operative recovery was uneventful, and a follow-up chest CT scan was normal. Histopathology report showed numerous small cystic structures lined by ciliated columnar cuboidal and columnar epithelium and fibromuscular layer consistent with CCAM.
CTEPH should be considered if dyspnea persists for more than 3 months after a confirmed PE diagnosis [9]. When CTEPH is suspected, a stepwise evaluation and management are needed.
If the patient has signs and symptoms of pulmonary hypertension despite 3 months of anticoagulation following acute PE, Transthoracic Echocardiogram and 6 minutes walk test should be done. Echocardiographic findings are right ventricular dilatation resulting in tricuspid regurgitation (TR) which presents as tricuspid valve peak systolic gradient of more than 60 mm Hg, if evidence of pulmonary hypertension is evident, the next step is a V/Q scan. A negative V/Q can effectively rule out pulmonary embolism, V/Q scintigraphy is more sensitive than CTAP in detecting CTEPH [10]. But V/Q scan has some limitations in determining the exact location, burden, and extent of pulmonary embolism which are very important information in figuring out the appropriate management plan. So, if a mismatched segmental defect is noticed in the V/Q scan, computed tomographic angiography is done for further evaluation. CT-AP reliably identifies major vessels clot but it has limitations in diagnosing distal vessels clot. That’s why it is not the initial diagnostic tool for PE [10]. If computed tomographic angiography shows suspicion of pulmonary hypertension, the next step is to refer to the CTEPH center for right heart catheterization with pulmonary angiogram +/- CT angiogram to quantify the degree of pulmonary hypertension as well as to assess responsiveness to vasodilator. If vasodilators respond by resulting in a reduction of pulmonary artery pressure, it indicates that for the patient who undergoes pulmonary endarterectomy, the long-term survival rate will increase [11].
Once the disease is confirmed, the patient showed be assessed for operability. Right heart catheterization and CT pulmonary angiography are the gold standards for the assessment of operability [12]. If the patient is a Candidate for surgery, pulmonary endarterectomy is the treatment of choice. Even though it is a complicated procedure, it increases the quality of life of the patient. Studies have shown that after a successful thromboendarterectomy, the patient’s condition improves significantly resulting in decreasing pulmonary vascular resistance and as a result improving cardiac output [13]. Complication after PEA includes reperfusion pulmonary edema, pulmonary artery steal syndrome, residual pulmonary hypertension, and recurrent thromboembolism [14].
Pharmacological management +/-balloon angioplasty should be offered for patients who are not candidates for surgery or recurrent pulmonary embolism despite PEA.
Pharmacological management consists of diuretics, Riociguat, lifelong anticoagulation therapy, and oxygen therapy for heart failure and/or hypoxemia [15]. Endothelin receptor antagonists such as Ambrisentan and bosentan improve exercise tolerance in patients with PAH [16]. Studies showed Riociguat was effective and well-tolerated in patients of advanced age or risk factors for HFrEF. The long-term extension study demonstrated that the use of Riociguat is safe to use for up to 3 years after initiation [17]. Adverse effects of RIociguatincludes hypotension, dyspnea, dyspepsia, headaches, and upper respiratory tract infection. Macitentan also showed benefits in decreasing PVR. Epoprostenol is also very effective as it works as a pulmonary and systemic vasodilator, resulting in decreasing pulmonary vascular resistance and increasing oxygen delivery to the body [18,19]. In cases of inoperable CTEPH, Balloon pulmonary angioplasty is a promising alternative to PEA [20]. Balloon pulmonary angioplasty (BPA) has shown some benefits in both hemodynamic and functional improvement [21]. It improves right ventricular function, causes myocardial remodeling, and alleviates dyssynchrony, as well as improves left ventricular function. By improving pulmonary vascular resistance, BPA helps in the reduction of troponin-T levels, suggesting attenuation of myocardial injury. BPA also helps in improving respiratory function and oxygenation [22,23]. Even though normal pulmonary function cannot be restored fully, early treatment can prevent permanent damage to pulmonary vasculature and thus prevent the development of decompensated heart failure.
The exact etiology of CCAM remains unknown. There is a hypothesis that hamartomata’s change in the terminal bronchioles or an arrest in their embryological development between 7 and 15 weeks of gestation is the suspected cause [7,8]. Another theory is that decreased apoptosis also plays a role; genetic studies showed that the related genes in the pathogenesis are HOXB5, Fgf7, and PDGFB [9].
Although congenital cyst adenomatoid malformation, also known as congenital airway malformation, is a rare disease but still, it is the most common malformation of the lower respiratory tract. In CCAM, the normal alveoli are replaced by a cystic component composed of adenomatous hyperplastic bronchioles. Due to recent advances in prenatal care and ultrasound, cystic lung lesions are more often detected early, and accordingly, peri Partum and neonatal care are planned. Stocker classified CCAM into five types from 0 to IV, as shown in the figure. Among all, Type III has the worst prognosis. CCAM is currently organized into five main types (0-4) based on the embryologic level of origin and the histologic features [10,11] (Figures 1-3, Table 1).
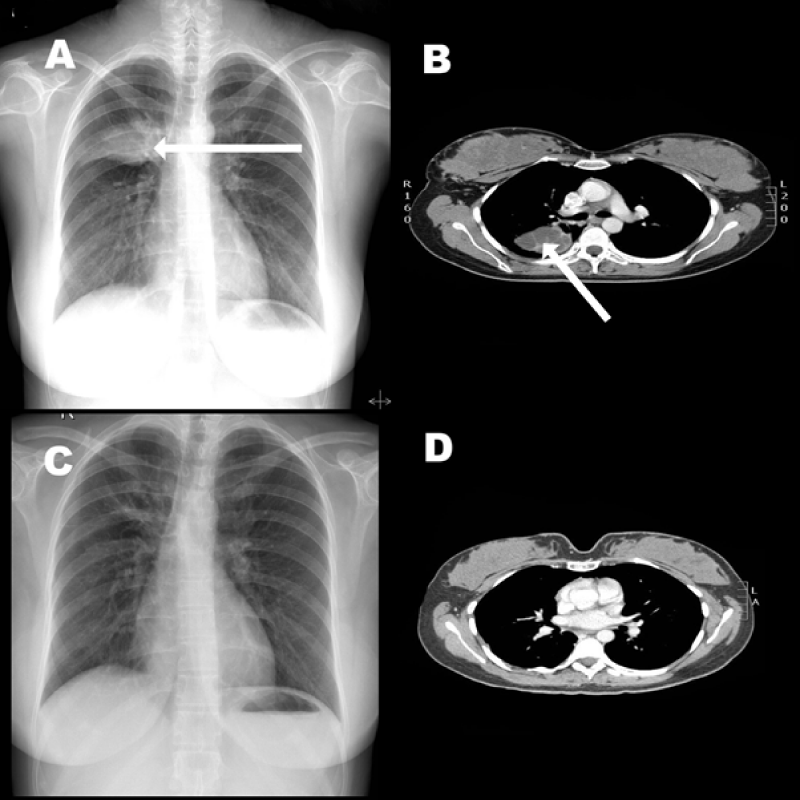
Figure 1: (A) Chest X-Ray showing a opacity in the right lung upper zone. (B) CT scan of the chest showing heterogenic mass in the right lung. (C) A normal post-operative chest X-Ray (D) Follow up CT scan of the chest revealed no abnormality.
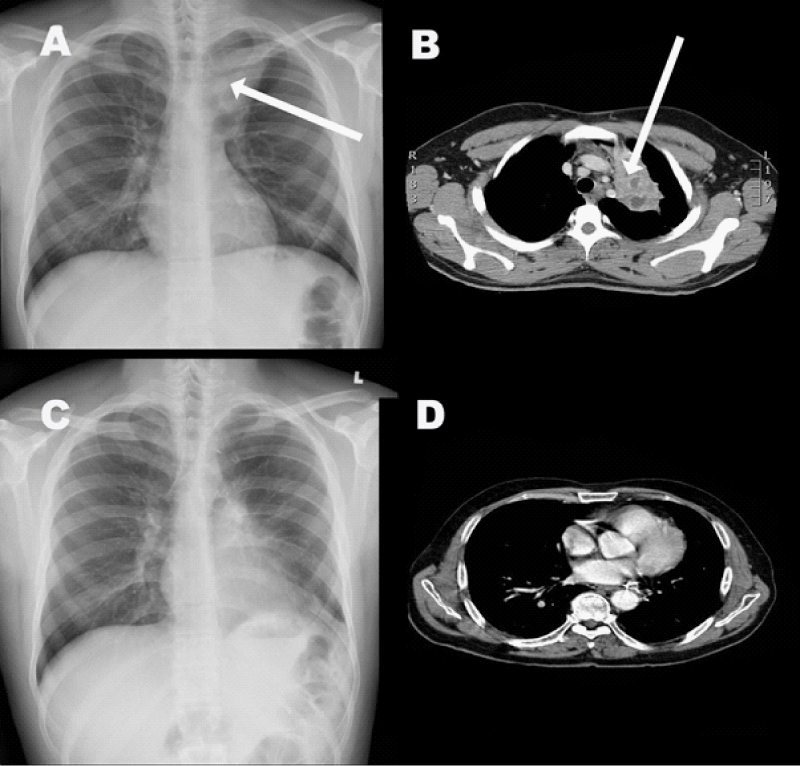
Figure 2: (A) Chest X-Ray showing a lesion in the upper lobe of left lung. (B) CT Scan of the chest showing complex cystic mass in the left upper lung. (C) A post-operative chest X-Ray. (D) Follow up CT scan of the chest.
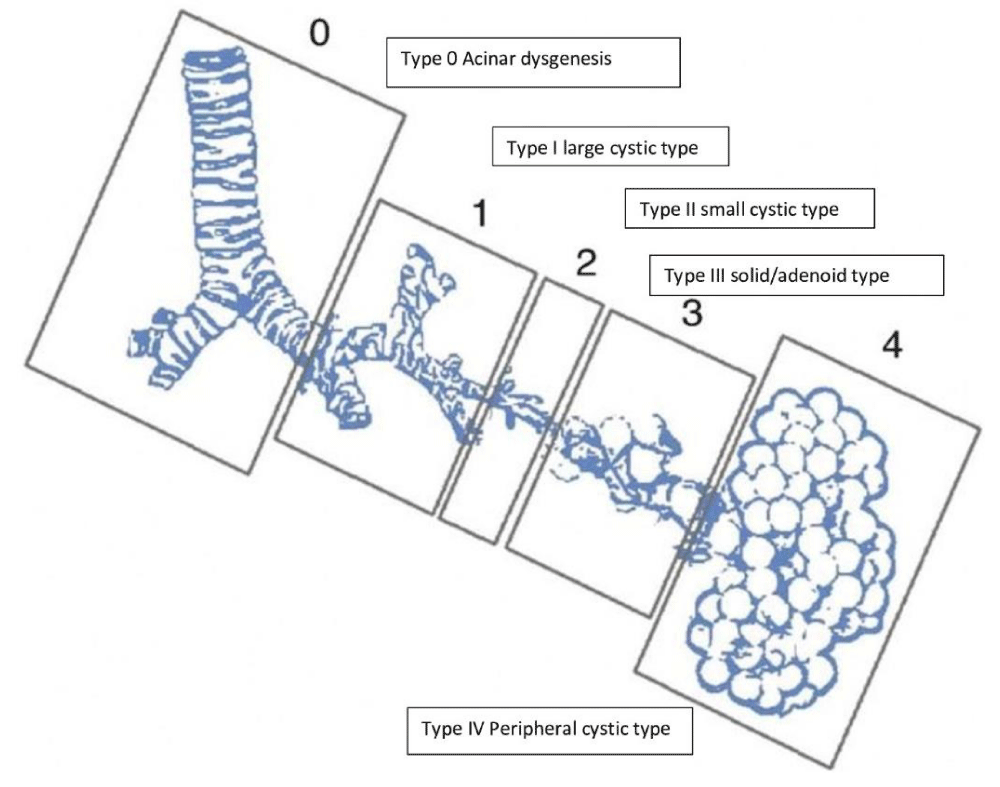
Figure 3: JT Stocker, et al. Pediatric pathology. Stoker classification with expanded form.
| Table 1: Classification of congenital Cyst adenoid malformation (CCAM). | |
| Type 0 | Type 0: CCAM is the rarest form and arises from the trachea or bronchus; there is acinar dysgenesis, and cysts are small |
| Type 1 | CCAM is the most common form, representing 50% to 70% of cases, and it arises from the distal bronchus or proximal bronchiole. Because these CCAMs may be large, they may have a significant mass effect, which can lead to hydrops. |
| Type II | Type II CCAMs account for 15% to 30% of cases and arise from terminal bronchioles. They are composed of smaller cysts, measuring 0.5 cm to 2 cm, and solid areas that may be difficult to distinguish from surrounding tissue. |
| Type III | CCAMs account for 5% to 10% of cases, are thought to arise from acinar-like tissue, and are composed of cysts so small the mass appears to be solid and highly echogenic on ultrasound. The tissue is acinar and shows adenomatoid elements consistent with the distal airway |
| Type IV | Type IV: CCAMs arise from alveoli accounting for 5% to 15% of cases. These CCAMs contain large peripheral cysts that may be as large as 10 cm and have been associated with malignancy, specifically pleuropulmonary blastoma. |
Type 0: CCAM is the rarest form and arises from the trachea or bronchus; there is acinar dysgenesis, and cysts are small.
Antenatally, CCAMs have been classified as microcystic (5 mm) versus macrocytic (.5 mm). Microcystic lesions are usually significantly larger. Laberge, et al. l and stoker, et al. l; reported Incidence of CCAM is 1 in 11,000 to I in 35000, respectively7. Usually, 25% of cases of CCAM are diagnosed during prenatal ultrasound check-ups. 85% are diagnosed in the first two years of life. At the same time, 71% are asymptomatic, which undergoes spontaneous regression [12].
CCAM in adults is very rare, and only fifty cases have been reported in the medical literature. CCAM is detected incidentally during the radiological examination, and patients are asymptomatic. Sometimes they present with chronic cough, recurrent respiratory tract infection, hemoptysis, pneumothorax, pneumonia, and lung abscess [13]. Medical literature shows that CCAM types I, II, and III are seen at 70%, 40% and 3% of the cases, respectively. Up to 26% of the patients were associated with other anomalies, more with type II and III, including abnormalities in chromosome 18 [14]. CCAM is sometimes misdiagnosed as sequestration as they share some histological patterns, but the criteria can differentiate them [15]. As given in Table 1. CCAM is usually diagnosed in the first two years of life, and around 17% of cases are present in the later years of life. It is commonly presented as a unilateral lesion causing respiratory distress in neonates and infants. If the fetus doesn’t develop hydrops, then postnatal survival is 100%. In adults, either this is usually diagnosed incidentally, rarely patients can present with a history of cough, shortness of breath, hemoptysis, pneumonia, and bacterial and fungal lung abscess. The best treatment for CCAM is surgical resection, lobectomy, or segmentectomy because there is a risk of malignant transformation if left untreated. CCAM is commonly isolated, but this has been reported in conjunction with cardiac and renal anomalies of 15% and 17%, respectively. Particularly type 2 is associated with gastrointestinal atresia and cardiac and renal skeletal anomalies [16,17]. CAM and extrapulmonary sequestration can occur together. Transitions into bronchioloalveolar carcinoma, pleuropulmonary blastoma and rhabdomyosarcoma have been reported [18-20].
CT scan chest is the diagnostic imaging tool for CCAM, but histological confirmation is mandatory; therefore, diagnosis is reached only from the histopathology post-surgical resection of the lesion. The standard treatment of CCAM is lobectomy because of poor sensitivity and low negative predictive value of the preoperative CT, which decreases the chance of recurrence [20]. There are conditions when CCAM with extensive involvement and difficult surgical technique are treated conservatively. The operative surgical versus nonoperative treatment decision depends on the extent of the disease, patient symptoms, and risk for surgery. We recommend early surgical intervention in an asymptomatic patient who is fit for surgery because it will be easier than when it becomes symptomatic and infected. There is always a risk of complications in CCAM like pneumothorax, infection, and malignancy transformation [21-24].
- Harmath A, Csaba A, Hauzman E, Hajdú J, Pete B, Papp Z. Congenital lung malformations in the second trimester: prenatal ultrasound diagnosis and pathologic findings. J Clin Ultrasound. 2007 Jun;35(5):250-5. doi: 10.1002/jcu.20341. PMID: 17373682.
- CH'IN KY, TANG MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol (Chic). 1949 Sep;48(3):221-9. PMID: 18137795.
- Stocker JT. Congenital pulmonary airway malformation: a new name for an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology 2002; 41: 424–458.
- MacSweeney F, Papagiannopoulos K, Goldstraw P, Sheppard MN, Corrin B, Nicholson AG. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol. 2003 Aug;27(8):1139-46. doi: 10.1097/00000478-200308000-00012. PMID: 12883247.
- Adzick NS, Harrison MR, Glick PL, Golbus MS, Anderson RL, Mahony BS, Callen PW, Hirsch JH, Luthy DA, Filly RA, et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg. 1985 Oct;20(5):483-8. doi: 10.1016/s0022-3468(85)80470-x. PMID: 3903097.
- Agha RA, Borrelli MR, Farwana R, Koshy K, Fowler AJ, Orgill DP; PROCESS Group. The PROCESS 2018 statement: Updating Consensus Preferred Reporting Of CasE Series in Surgery (PROCESS) guidelines. Int J Surg. 2018 Dec;60:279-282. doi: 10.1016/j.ijsu.2018.10.031. Epub 2018 Oct 22. PMID: 30359781.
- Luján M, Bosque M, Mirapeix RM, Marco MT, Asensio O, Domingo C. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration. 2002;69(2):148-54. doi: 10.1159/000056318. PMID: 11961429.
- Volpe MV, Pham L, Lessin M, Ralston SJ, Bhan I, Cutz E, Nielsen HC. Expression of Hoxb-5 during human lung development and in congenital lung malformations. Birth Defects Res A Clin Mol Teratol. 2003 Aug;67(8):550-6. doi: 10.1002/bdra.10086. PMID: 14632303.
- Jancelewicz T, Nobuhara K, Hawgood S. Laser microdissection allows detection of abnormal gene expression in cystic adenomatoid malformation of the lung. J Pediatr Surg. 2008 Jun;43(6):1044-51. doi: 10.1016/j.jpedsurg.2008.02.027. PMID: 18558180.
- Stocker JT. The respiratory tract. In: Stocker JT, Dehner LP, eds. Pediatric pathology. Philadelphia, PA: Lippincott Williams & Wilkins.2001; 445-517.
- Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003 Feb;12(1):17-37. doi: 10.1053/spsu.2003.00001. PMID: 12520470.
- Laberge JM, Flageole H, Pugash D, Khalife S, Blair G, Filiatrault D, Russo P, Lees G, Wilson RD. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther. 2001 May-Jun;16(3):178-86. doi: 10.1159/000053905. PMID: 11316935.
- Zach MS, Eber E. Adult outcome of congenital lower respiratory tract malformations. Thorax. 2001 Jan;56(1):65-72. doi: 10.1136/thorax.56.1.65. PMID: 11120908; PMCID: PMC1745897.
- Cloutier MM, Schaeffer DA, Hight D. Congenital cystic adenomatoid malformation. Chest. 1993 Mar;103(3):761-4. doi: 10.1378/chest.103.3.761. PMID: 8449065.
- Gornall AS, Budd JL, Draper ES, Konje JC, Kurinczuk JJ. Congenital cystic adenomatoid malformation: accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn. 2003 Dec 15;23(12):997-1002. doi: 10.1002/pd.739. PMID: 14663837.
- Collins AM, Ridgway PF, Killeen RP, Dodd JD, Tolan M. Congenital cystic adenomatoid malformation of the lung: hazards of delayed diagnosis. Respirology. 2009 Sep;14(7):1058-60. doi: 10.1111/j.1440-1843.2009.01603.x. PMID: 19740267.
- Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009 Jan;44(1):14-30. doi: 10.1002/ppul.20917. PMID: 19061226.
- Laje P, Liechty KW. Postnatal management and outcome of prenatally diagnosed lung lesions. Prenat Diagn. 2008 Jul;28(7):612-8. doi: 10.1002/pd.1966. PMID: 18330859.
- West D, Nicholson AG, Colquhoun I, Pollock J. Bronchioloalveolar carcinoma in congenital cystic adenomatoid malformation of lung. Ann Thorac Surg. 2007 Feb;83(2):687-9. doi: 10.1016/j.athoracsur.2006.06.029. PMID: 17258019.
- Dahabreh J, Zisis C, Vassiliou M, Arnogiannaki N. Congenital cystic adenomatoid malformation in an adult presenting as lung abscess. Eur J Cardiothorac Surg. 2000 Dec;18(6):720-3. doi: 10.1016/s1010-7940(00)00578-9. PMID: 11113682.
- Khan NU, Jones MT, Greaves M. Case report: Congenital cystic adenomatoid malformation of an entire lung in a 33-year-old man: a case report and review of the literature. Br J Radiol. 2008 Nov;81(971):e276-8. doi: 10.1259/bjr/23523404. PMID: 18941042.
- d'Agostino S, Bonoldi E, Dante S, Meli S, Cappellari F, Musi L. Embryonal rhabdomyosarcoma of the lung arising in cystic adenomatoid malformation: case report and review of the literature. J Pediatr Surg. 1997 Sep;32(9):1381-3. doi: 10.1016/s0022-3468(97)90329-8. PMID: 9314270..
- Hulnick DH, Naidich DP, McCauley DI, Feiner HD, Avitabile AM, Greco MA, Genieser NB. Late presentation of congenital cystic adenomatoid malformation of the lung. Radiology. 1984 Jun;151(3):569-73. doi: 10.1148/radiology.151.3.6718709. PMID: 6718709..
- Muller CO, Berrebi D, Kheniche A, Bonnard A. Is radical lobectomy required in congenital cystic adenomatoid malformation? J Pediatr Surg. 2012 Apr;47(4):642-5. doi: 10.1016/j.jpedsurg.2011.08.002. PMID: 22498375.