
More Information
Submitted: July 21, 2021 | Approved: August 04, 2021 | Published: August 05, 2021
How to cite this article: Nieves-Ortiz AA, Hernandez-Moya K, Garcia-Puebla J, Padilla-Rodriguez K, Roman-Velez N, et al. “Fatty Lungs”: An uncommon case of Autoimmune Pulmonary Alveolar Proteinosis. J Pulmonol Respir Res. 2021; 5: 076-077.
DOI: 10.29328/journal.jprr.1001029
Copyright License: © 2021 Nieves-Ortiz AA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Pulmonary alveolar proteinosis; GM-CSF autoantibodies; Autoimmune

“Fatty Lungs”: An uncommon case of Autoimmune Pulmonary Alveolar Proteinosis
Arnaldo A Nieves-Ortiz1*, Kyomara Hernandez-Moya1, Juan Garcia-Puebla2, Kimberly Padilla-Rodriguez2, Neshma Roman-Velez1, Giovanni Veloz-Irizarry2, Keren Mendez-Ramirez2, Hector Collazo-Santiago2, Jose Adorno-Fontanez2 and Ricardo Fernandez-Gonzalez2
1Internal Medicine Department, San Juan City Hospital, San Juan, 00935 Puerto Rico
2Pulmonary Disease and Critical Care Medicine Department, San Juan City Hospital, San Juan, 00935 Puerto Rico
*Address for Correspondence: Arnaldo A Nieves-Ortiz, MD2, HC-2 Box 6558, Bo. Campo Rico, Canovanas, 00935 Puerto Rico, Tel: 787-423-2261; Email: [email protected]
Pulmonary Alveolar Proteinosis (PAP) is a rare lung disease characterized by excessive accumulation of surfactant lipids and proteins in alveoli and terminal airways. It is caused by impaired GM-CSF signaling [1]. Surfactant is synthesized and secreted by alveolar type II epithelial cells, and removed by uptake and catabolism by these cells, and the alveolar macrophages. Patients with PAP usually describe gradual onset of progressive exertional dyspnea and non-productive cough. However, an asymptomatic presentation is observed in up to 25% of cases, even in the presence of diffuse radiographic changes. Three recognized subtypes exist. Autoimmune PAP is associated with neutralizing GM-CSF autoantibodies and accounts about 90% of cases. Secondary PAP may occur in the context of any disease that reduces the abundance or functionality of alveolar macrophages, resulting in impaired surfactant clearance. Congenital PAP is the result of genetic mutations that disrupt GM-CSF signaling, including mutations in the α- or β-chains of the GM-CSF receptor [1-3].
PAP occurs approximately 2-3 times more commonly in men, at a median age of 35-51 years. The prognosis for autoimmune PAP is variable and may involve spontaneous resolution (reported in 5% - 25%); persistent, unremitting symptoms and/or progressive disease leading to respiratory failure.
A 32 y/o female patient with medical history of asthma, diabetes mellitus type 2, morbid obesity, papillary thyroid CA and hypothyroidism who presented to the urgency room due to worsening dyspnea on exertion and dry cough for the past 6 months ago. She denied night sweats, hemoptysis, chest pain, or weight changes. Outpatient medications included montelukast, ipratropium and albuterol inhaler, fluticasone/vilanterol, metformin and levothyroxine. She denies smoking or any other significant environmental exposure. Vitals signs were remarkable for tachycardia and desaturation. Physical Examination remarkable for decrease breath sounds. ABG remarkable for severe hypoxemia with elevated A-a gradient. CBC remarkable for leukocytosis with neutrophilia and lymphopenia. Chemistry was remarkable with increased lactate dehydrogenase. Inflammatory markers including CRP and ESR, anti-thyroid peroxidase, thyroglobulin and rheumatologic workup were unremarkable. Chest X-Ray showed bilateral perihilar, infrahilar and basilar opacities. Chest CT shows lung parenchyma with increased interstitial markings as well as peribronchial cuffing, diffuse ground-glass and confluent densities with crazy paving pattern. Anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) antibodies was positive. Our patient was treated with whole-lung lavage (WLL) and GM-CSF therapy with excellent clinical and radiological improvement. In the first left WLL, the process was repeated for 9 cycles until the effluent became clear for a total instilled volume of 7 liters. One week later, we performed the right WLL and the process was repeated for 19 cycles until the effluent became clear for a total instilled volume of 8.5 liters (Figures 1-5).
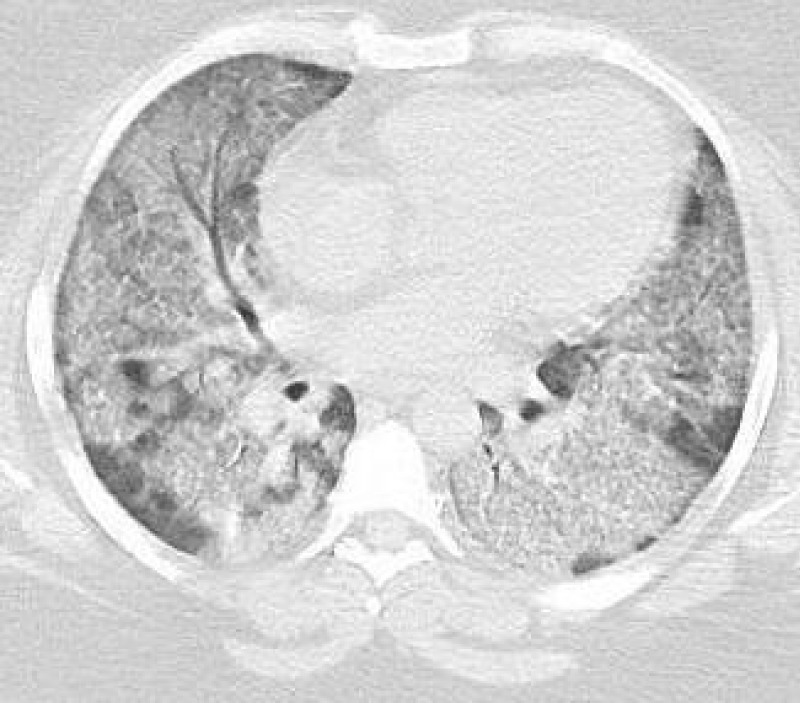
Figure 1: Pre WLL. Lung parenchymal shows increased interstitial markings as well as peribronchial cuffing, diffuse ground glass and confluent densities with crazy paving pattern.
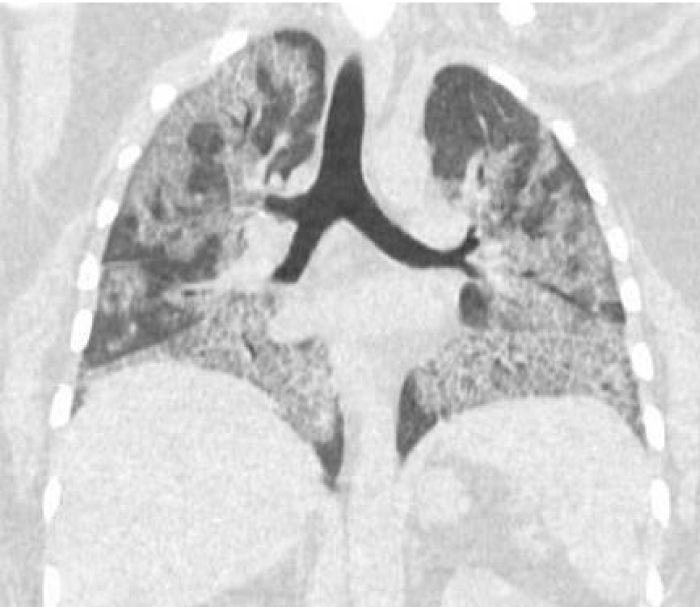
Figure 2: Pre WLL. Lung parenchymal shows increased interstitial markings as well as peribronchial cuffing, diffuse ground glass and confluent densities with crazy paving pattern.
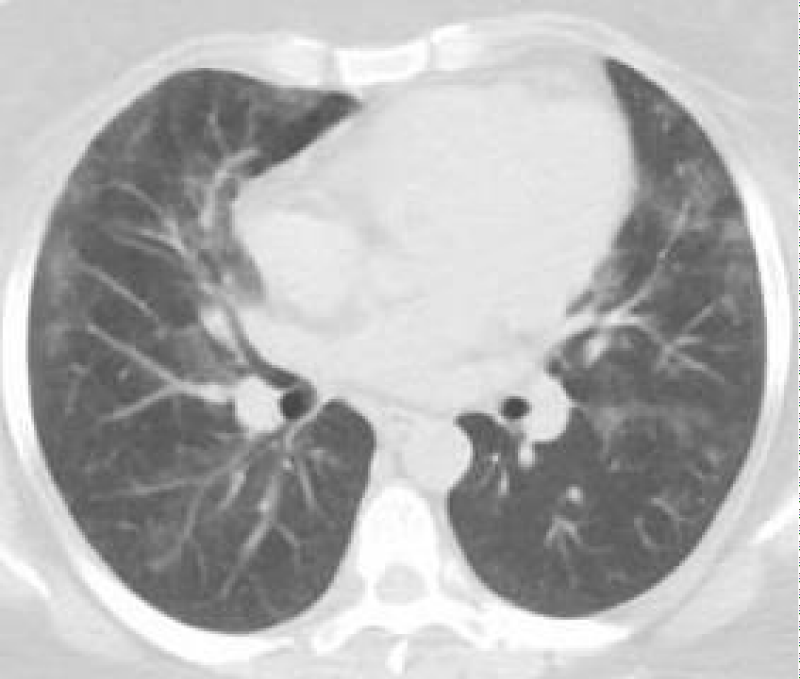
Figure 3: Post WLL. Lung parenchyma shows improvement from previous CT after WLL.
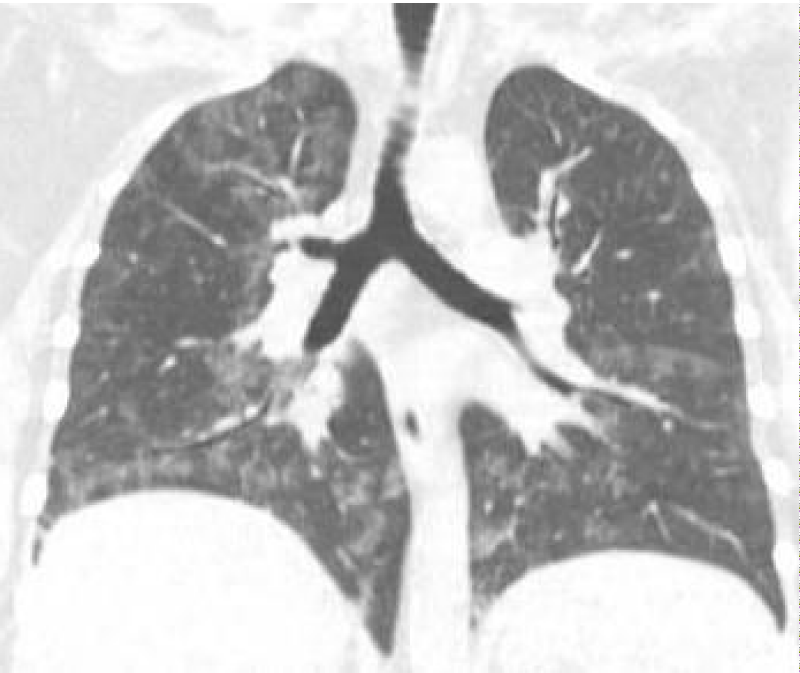
Figure 4: Post WLL. Lung parenchyma shows improvement from previous CT after WLL.

Figure 5: Collection canister from WLL fluid with progressive translucency of fluid from 1 through 4.
PAP continue to be a very rare condition with an incidence of 6-7 cases per million in the general population. Symptoms are non-specific, and includes dyspnea on exertion along with productive cough with whitish sputum. Gold standard diagnosis is the anti GM-CSF antibodies and bronchoscopy with proteinaceous material. WLL remains the standard of treatment with over 70% of successful response. With our case we expect to provide awareness and knowledge to the medical field to always include PAP in the working diagnosis of all patients with progressive dyspnea. There are available diagnostic tools at our hands that can be promptly used to increase the rate of diagnosis of this syndrome, with the expected improvement in the quality of life of our patients.
We will like to acknowledge the department of Pulmonary and Critical Care Fellowship Program, San Juan City Hospital. We have no conflict of interest to declare. This statement is to certify that all authors have seen and approved the manuscript being submitted.
- Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003; 349: 2527-2539. PubMed: https://pubmed.ncbi.nlm.nih.gov/14695413/
- Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med. 2016; 37: 431-440. PubMed: https://pubmed.ncbi.nlm.nih.gov/27514590/
- DynaMed. Pulmonary Alveolar Proteinosis (PAP). EBSCO Information Services. 2021. https://www.dynamed.com/condition/pulmonary-alveolar-proteinosis-pap